![]()
|
|---|
| Conférences | |||||
|---|---|---|---|---|---|
 |
Cette page est en construction. |
Spectroscopie hautement résolue du dimere (HF)2 et ses isotopomères comme prototype de système à liaison hydrogène
Dans cette étude, le dimère (HF)2 est utilisé comme prototype pour mieux comprendre et décrire les dynamiques de systèmes liés par liaison hydrogène. La grande simplicité de ce dimère nous permet d'utiliser la spectroscopie rovibrationelle hautement résolue ainsi qu'un potentiel d'hypersurface 6-dimensionnel d'une très grande précision.
Le temps de prédissociation (tPD) du dimère est spécifique au mode de vibration excité. Dans la région des modes d'étirement de (HF)2, des temps de vie ont été mesurés dans un jet moléculaire par specroscopie à résolution sub-Doppler: l'excitation vibrationnelle du mode d'étirement impliqué dans la liaison hydrogène (nb) conduit à un temps de prédissociation plus de 20 plus court que celle du mode d'étirement "libre" non impliqué dans la liaison hydrogène (nf). Cette sélectivité de modes est préservée dans la polyade N=2 ce qui correspond à l'excitation de deux quanta de modes d'étirement de HF, entre 7500 et 7800 cm-1, i.e. plus de sept fois l'énergie nécessaire à la dissociation de la liaison hydrogène. Les trois différentes excitations conduisent à des dynamiques de dissociation différentes: dans notre laboratoire, des mesures par spectroscopie IRTF ont révélé un comportement contraire à RRKM. Plus récemment, des temps de prédissociation ont été mesurés par une expérience de cavity-ring-down hautement résolue combinée à un jet moléculaire: le temps de prédissociation suite à l'excitation du mode d'étirement impliqué dans la liaison hydrogène est beaucoup plus court que celui suite à l'excitation du mode d'étirement libre.
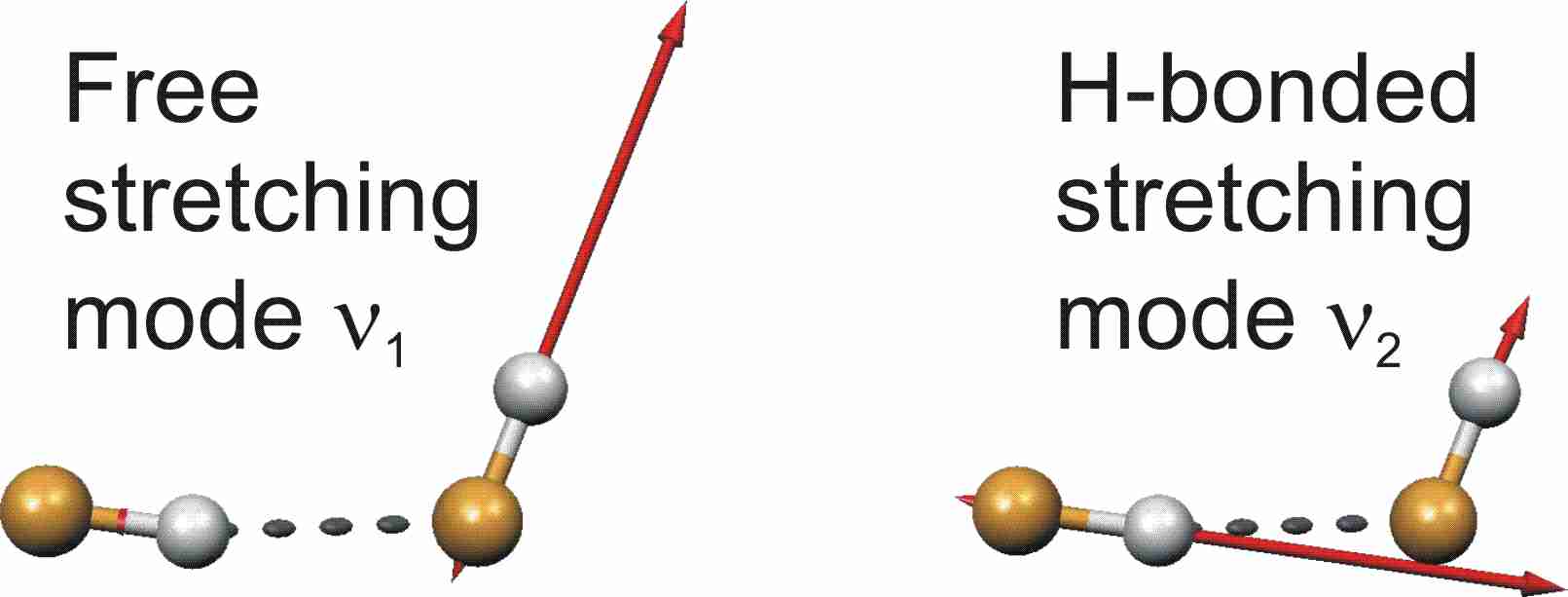
Nous avons réoptimisé le système expérimental composé d'un jet supersonique couplé à la spectroscopie CRD. Cette optimisation nous permet désormais de travailler avec un jet moléculaire plus froid qu'auparavant. Nous avons aussi mesuré les temps de prédissociation des isotopomères HF·DF and DF·HF.
Parallèlement aux mesures expérimentales, nous avons réalisé des calculs de dynamique moléculaire classique sur le dimère (HF)2 dimer avec les surface d'énergie potentientelle 6-dimensionnelles SO-3 et SQSBDE pour estimer le temps de prédissociation tPD pour plusieurs excitations initiales. C'est la première prédiction de tPD pour des excitations comprenant deux ou trois quanta de vibration d'élongation HF. Nos calculs reproduisent sélectivité de modes dans les valeurs de tPD observée experimentallement. Les excitations comprenant le mode d'élongation impliquant la liaison hydrogène génère des temps de prédissociation plus courts que celles comprenant le mode d'élongation libre. Nous étudions aussi l'influence des modes d'élongation intermoléculaire et de déformation sur tPD. De plus, nos spectres calculés sont en bon accord avec les spectres d'absorption infrarouge, au niveau des positions de bandes et des intensités relatives. Cette étude fournit non seulement des informations pour le dimère de HF, mais aussi un système de référence pour les calculs de type classique.
Etude du spectre de méthane dans la region de l'icosade
Nous avons réalisé une étude en fonction de la temperature du spectre rovibrationel des deux principaux isotopomères du méthane (12CH4 et 13CH4) dans la région de icosade. Notre instrument jet - CRDS nous permet de refroidir les jet supersoniques jusu'à des températures rotationelles de 4 K et de réaliser des mesures semi-quantitatives.
La comparaison des spectres enregistrés at différentes températures rotationelles entre 4 K et 300 K permet de proposer une attribution univoque des branches Q et R de la band de combinaison n2 + 2n3. Nous avons résolu des contradictions au niveau de l'attribution pour 12CH4 et présentons une attribution définitive pour les lignes correspondant à un nombre quantique de moment angulaire jusqu'à J = 4.
Consevation ou relaxation de spin nucléaire dans des petites molécules
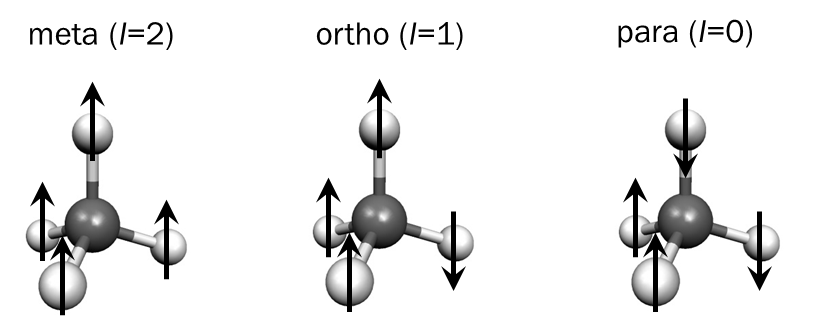
Les molécules avec plusieurs noyaux identiques ayant un spin nucléaire non nul existent sous plusieurs forms appelées isomères de spin nucléaire; par exemple, le méthane (CH4), l'ammoniac (NH3) ou l'eau (H2O) sont des candidats possibles pour explorer la conservation ou conversion de symmetry de spin nucléaire. Si la symmétrie de spin nucléaire est conservée au cours d'un processus de refroidissement, les isomères de spin nucléaires gardent leurs populations établies avant le refroidissement et on observe une superposition de plusieurs distributions selon la loi de Boltzmann. Par contre, en cas de relaxation de symmétrie de spin nucléaire, les états despin peuvent changer et on observe un équilibre thermique global sur tous les états.
Nous avons d'abord utilisé le méthane pour étudier la conservation de symmétrie de spin nucléaire en jet supersonique avec notre instrument jet-CRDS. L'analyse des intensités relatives des spectres enregistrés à des températures rotationnelle entre 50 K et moins que 10 K indique une conservation de symmétrie de spin nucléaire lors de l'expansion supersonique; ceci est en accord avec les précédants résultats utilisant d'autres techniques ainsi qu'avec les théorie des jets moléculaires supersoniques.
L'eau est un candidat intéressant pour ce genre d'études: une relaxation de symmétrie de spin nucléaire serait intéressants par exemple pour les mesures interstellaires et a déjà été observée en matrices de gaz rares. Nous avons utilisé la même approche que pour le méthane pour étudier la symmétrie de spin nucléaire de l'eau en jet supersonique d'argon ou d'oxygène dans la région de la bande 2n3 et à des températures inférieures à 30 K. Pour les plus faibles concentrations d'eau utilisées dans l'expansion, nous avons observé une conservation de symmétrie de spin nucléaire, comme pour nos précédentes expériences avec le méthane. La relaxation de symmétrie de spin nucléaire que nous avons, de façon surprenante, à "forte" concentration d'eau dans l'expansion est expliquée par la formation de clusters d'eau. Des résultats similaires ont été observés pour des expansions d'eau dans l'oxygène au lieu de l'argon, ce qui ne révèle pas d'effet significatig du moment magnétique du partenaire de collision.

Redistribution intramoléculaire d'énergie vibrationnelle
Dans notre groupe, nous utilisons deux approches expérimentales pour étudier la redistribution intramoléculaire d'énergie vibrationnelle (IVR): l'approche résolue en temps (fs "pump-probe") et l'approche utilisant la spectroscopie à haute résolution, sans résolution temporelle.
Les isotopomères deutérés de l'iodure de méthyle (CHD2I et CH2DI) sont de bons candidats pour l'étude de la redistribution d'énergie vibrationnelle après excitation dans la région de l'harmonique des modes d'élongation CH vers les modes de basse fréquence impliquant le groupement CI. Des expériences utilisant l'instrument fs-pump-probe dans notre groupe ont montrés que CHD2I présente plusieurs temps de relaxation IVR (de fs à ps), ce qui révèle des mécanismes de couplage intramoléculaires différents. Parallèlement, notre travail utilisant la spectroscopie à moyenne résolution a mis en évidence un fort couplage de type Fermi entre les modes d'élongation et de déformation de CHD2I, qui conduit à des temps de redistribution trèS courts de l'ordre de 100 fs. La prochaine étape consiste en l'analyse rovibrationnelle des spectres à très haute résolution des bandes fondamentales enregistrées avec notre spectromètre infrarouge à transformée de Fourier: une étude systématique devrait permettre de mettre en évidence les couplages plus faibles qui existenent entre les différents modes et de comprendre les temps de redistribution plus longs observés dans les expériences fs Pump-Probe. Notre étude de la région du mode d'élongation n1 ainsi que celle de la région autour de 1000 cm-1 comprenant trois bandes fondamentales ont révélé des couplages faibles qui peuvent être reliés aux processus lents de redistribution d'énergie.
Transfert d'hydrogene/de proton le long d'une chaine de solvants liés par liaison hydrogene
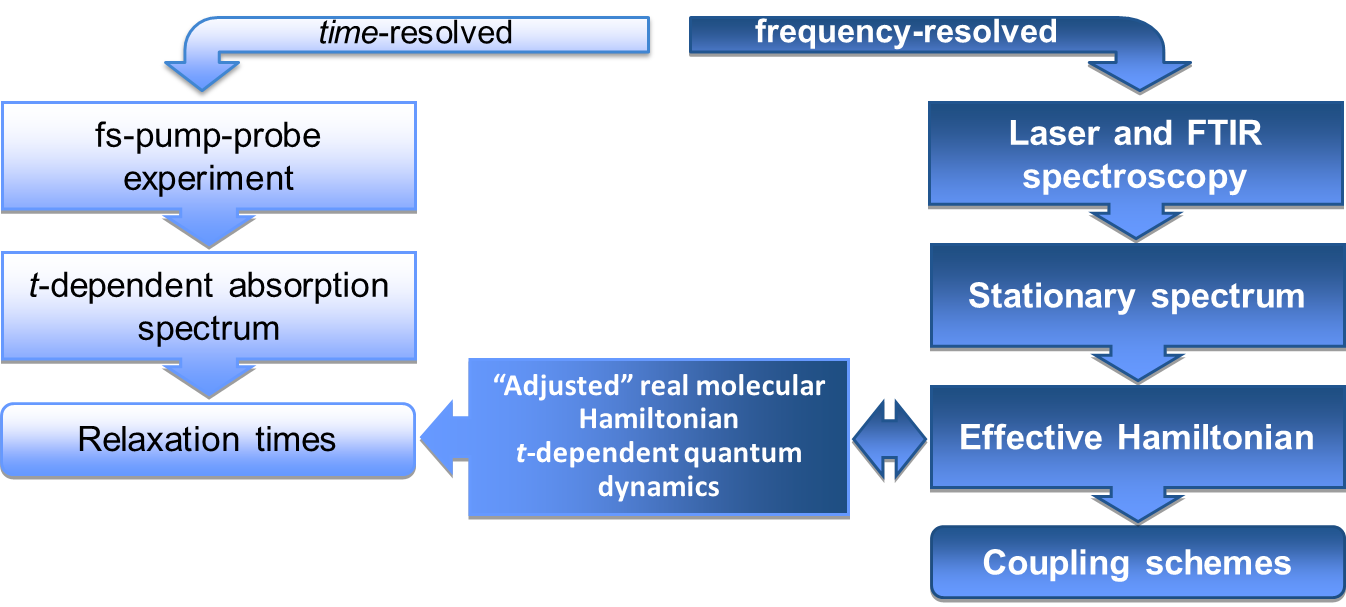
Les transferts de proton et d'hydrogène font partie des réactions fondaöentales les plus étudiées en biologie, spécialement le transfert d'ions à travers les membranes (chaînes de protons). La 7-hydroxyquinoline est une molécule aromatique qui possède une groupement OH donneur de H, et un un site accepteur d'H (l'atome N); Dans le cadre de notre travail cette molécule est utilisée comme support pour une chaine de liaisons hydrogène, the deux sites terminant la chaîne étant suffisament espacés sur la molécule.
Le cluster 7-hydroxyquinoline•(NH3)3 a été utilisé comme modèle pour étudier le transfert d'atome d'hydrogène à l'état excité (ESHAT) le long de la cha`ne liiée par liaisons hydrogènes NH3 • • • NH3 • • • NH3. L'excitation électronique du cluster en jet moléculaire ne produit aucune réaction, tandis que l'excitation des vibrations de la chaîne d'ammoniac induit une fluorescence jaune du à la forme excitée du cluster 7-ketoquinoline•(NH3)3, prouvant la tautomérisation énol-kéto à l'état excité. Les calculs ab initio confirment que la réaction se produit via des transferts d'hydrogène successifs le long de la chaîne d'ammoniac dans un processus dit de Grotthus; les mouvements du proton et de l'électron sont couplés. La barrière de la première étape du mécanisme provient d'un croisement entre deux états excités pp* et ps*, le deuxiéme étant de type Rydberg. Nous avons également observé que les modes intermoléculaires du système accéléraient la réaction de manière beaucoup plus efficace que les modes intramoléculaires. Des études sont en cours sur l'effet de solvant et de solvsatation.
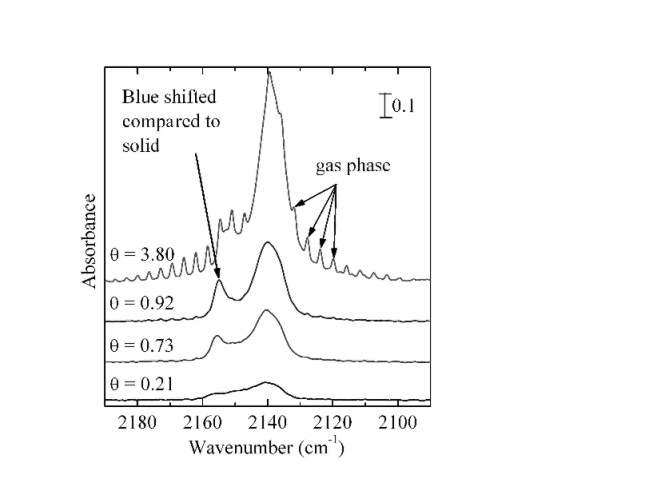
Physisorption sur de surfaces de glace amorphe (couplage entre la volumetrie isotherme d'adsorption et la spectroscopie infrarouge)
La physisorption sur des surfaces de glace amorphe entre 40 et 100 K est étudiée par couplage entre la manométrie isotherme d'adsorption et la spectroscopie IR-TF dans le cas de N2 CO, Ar, Kr, CH4 et CF4. Le couplage permet le tracé d' "isothermes infrarouge" qui a mis en évidence l'évolution de trois signaux associés à trois types de molécules de surface.

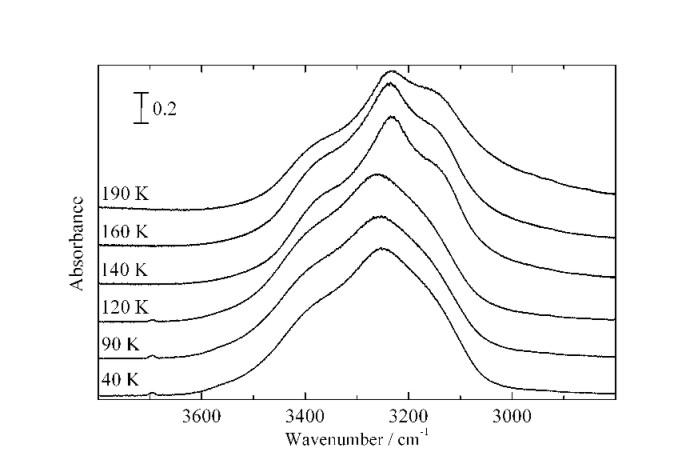
La glace amorphe formée présente une grande aire spécifique (supérieure a 100 m2g-1); cependant, l'analyse montre qu'elle n'est pas microporeuse.
L'étude comparative de l'adsorption des différents gaz a montré que leurs interactions avec la glace sont faibles, une liaison hydrogène étant identifiée dans le cas de N2 et CO ; la quantité adsorbée nécessaire pour recouvrir la surface est correlée au moment quadrupolaire de l'adsorbat. Des calculs quantiques de type Hartree-Fock périodiques ont mis en évidence un effet Stark vibrationnel expliquant le déplacement de la bande associée aux OH libres de surface.
Dans le cas de CO, une étude expérimentale approfondie en accord avec des calculs basés sur la théorie de la fonctionnelle de la densité a permis de compléter l'attribution vibrationnelle de CO adsorbé sur la glace amorphe et de modéliser trois sites d'adsorption ; elle a aussi montré l'existence d'interactions latérales stabilisantes de type L au sein de la couche adsorbée.
Les premiers résultats d'adsorption de N2 et CF4 sur la glace cristalline étudiée par spectroscopie d'absorption X sont présentés.
Réactivité/isomérisation induite par irradiations sélectives UV et IR
L'acétylacétone et la malonaldéhyde en matrices cryogéniques
Nous avons utilisé la malomaldéhyde et l'acétylacétone comme prototypes de systèmes à liaison hydrogène intramoléculaire. La forme chélatée de l'acétylacétone est la plus stable en matrice cryogénique. Des irradiations UV nous ont permis de mesurer les spectres UV des isomères non-chélatés à plus haute énergie et moins stables. Leur identification a été obtenue en comparant les spectres infrarouges induits par irradiations sélectives UV et IR avec des calculs de fréquences harmoniques ainsi que des calculs de transitions UV et de forces d'oscillateur.
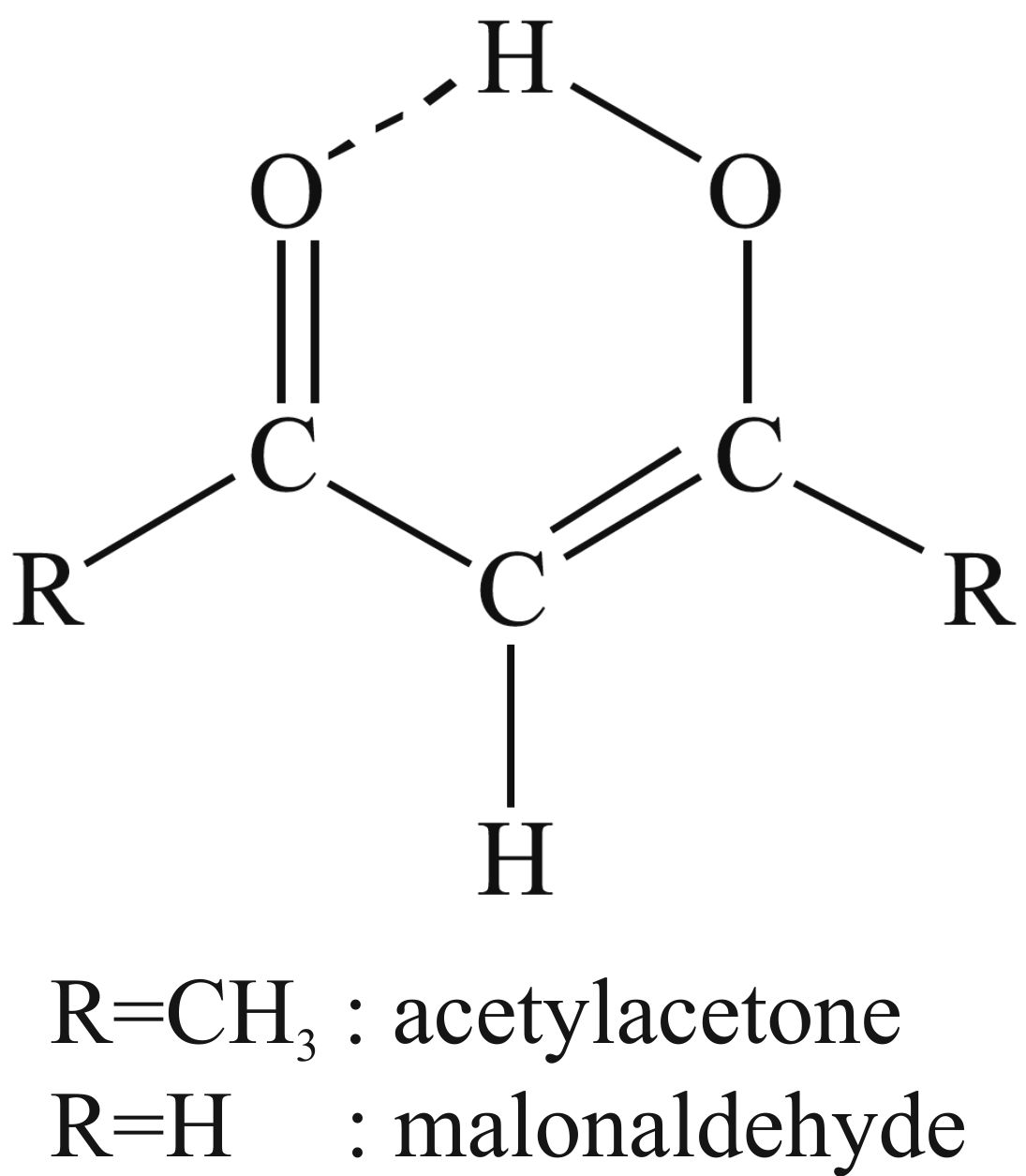
Nous avons également comparés la structure et la réactivité des huit isomères de la forme énol de l'acétylacetone et la malonaldehyde en utilisant à la fois des spectres expérimentaaux et des données théoriques. La force de la liaison hydrogène des formes chélatées est estimée grâce à la différence d'énergie entre la forme chélatée et les formes non-chélatées. Des irradiations infrarouge interconvertissent les isomères non-chélatés des deux molécules; l'efficacité et le taux de conversion dépend du gaz matriciel utilisé, ainsi que de la présence d'oxygène dans la matrice cryogénique, ce qui met en lumière une réactivité différente de celle de la phase gazeuse.
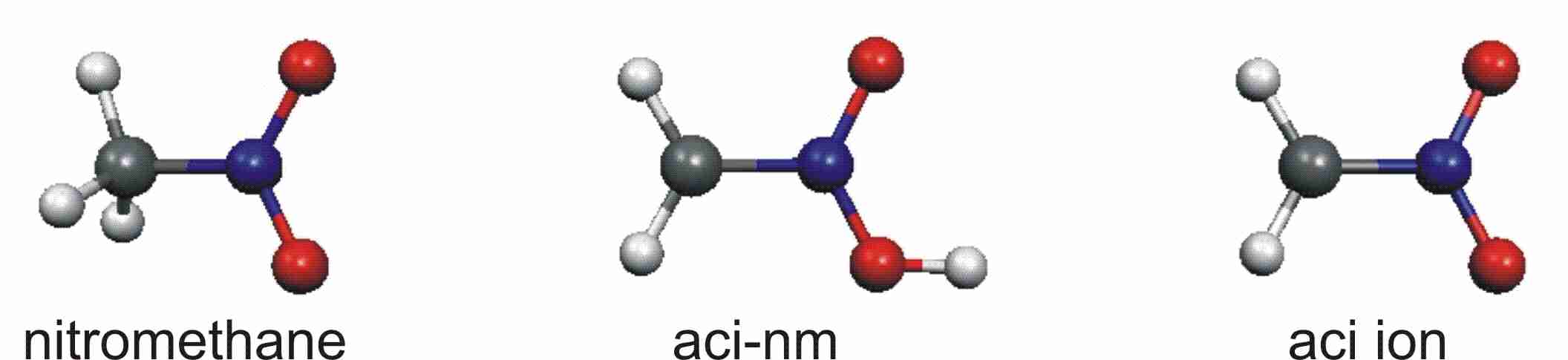
Le nitrométhane en matrices cryogéniques et phase condensée
Nous avons étudié la réactivité du nitrométhane induite par irradiation sélective. Cette étude a été réalisée dans le but de caractériser par spectroscopie IRTF les formes aci et aci ion susceptibles de se former après irradiation UV du nitrométhane et d'intervenir dans le processus de détonation. La dissociation de cette molécule n'est pas la même selon son environnement: Si en matrice cryogénique, la décomposition du nitrométhane conduit au méthylnitrite, c'est en phase ocndensée (liquide ou solide) que nous avons pu observer les deux formes recherchées.